 |
Case Report
A rare case of 21-hydroxylase deficiency presenting as precocious puberty in a male child
1 Chemical Pathologist, Department of Chemical Pathology, University of Limpopo, Polokwane, Limpopo Province, South Africa
2 Paediatrician Specialist, Department of Paediatrics, University of Limpopo, Polokwane, Limpopo Province, South Africa
3 Radiology Specialist, Department of Diagnostic Radiology, University of Limpopo, Polokwane, Limpopo Province, South Africa
4 Head of Department, Department of Paediatrics, University of Limpopo, Polokwane, Limpopo Province, South Africa
Address correspondence to:
Tumelo M Satekge
Chemical Pathologist, Department of Chemical Pathology, University of Limpopo, Polokwane, Limpopo Province,
South Africa
Message to Corresponding Author
Article ID: 100029Z19TS2025
Access full text article on other devices

Access PDF of article on other devices


How to cite this article
Satekge TM, Tshipeng JPM, Khosa RJ, Sutton C. A rare case of 21-hydroxylase deficiency presenting as precocious puberty in a male child. J Case Rep Images Pediatr 2025;7(2):5–9.ABSTRACT
Introduction: Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder that leads to impaired cortisol and/or aldosterone biosynthesis with simultaneous shunting to the intact androgen pathway. In contrast to females, males with CAH due to 21-hydroxylase deficiency may not always be detected at birth, and are often diagnosed later in life subsequent to life threatening adrenal crises and excessive virilization.
Case Report: Our case is a 9-year-old boy who presented at the age of 4 years with tall stature, a large penis, pubic hair as well as mild intellectual disability. His biochemical results revealed low follicle-stimulating hormone (FSH) and luteinizing hormone (LH) (<1.0 IU/L for both), elevated testosterone 12.5 nmol/L (0.1–0.9), and elevated 17-OH progesterone 87.0 nmol/L (0.5–2.2). Radiological imaging showed physeal closure in consonance with the age of 17 years, normal appearance of the adrenal glands on magnetic resonance imaging (MRI), however, adrenal rests were detected in his testes. The diagnosis was confirmed by biallelic pathogenic variants namely, c.293-13C>G splice site variant and c.955G>A p.(Gln319Ter) in the CYP21A2 gene.
Conclusion: The unavailability of universal newborn screening (NBS) in the low-and-middle-income countries contributes to treatable conditions such as CAH being missed particularly in male patients leading to potentially dire consequences to affected patients and their families. Comprehensive clinical assessment, together with appropriate laboratory and radiological investigations are fundamental for timely diagnosis and treatment of non-classical CAH.
Keywords: Congenital adrenal hyperplasia, CYP21A2 gene, 21-Hydroxylase deficiency, Precocious puberty
Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders encompassing enzyme deficiencies in the adrenal steroidogenesis pathway that lead to impaired cortisol and/or aldosterone biosynthesis with simultaneous shunting to the intact androgen pathway. Presentations vary from neonatal salt wasting and atypical genitalia, to adult presentation of hirsutism and irregular menses. Screening of neonates with 17-hydroxyprogesterone concentrations for classic (severe) 21-hydroxylase deficiency, the most common type of CAH, is in place in many countries. Long-term complications of CAH include abnormal growth and development, adverse effects on bone, the cardiovascular system and infertility [1]. Female neonates with CAH can be diagnosed early due to genital ambiguity contrary to male newborns whereby CAH due to 21-hydroxylase deficiency may not always be detected at birth and is diagnosed after a life-threatening adrenal crisis, excessive virilization or even after an unsuspected death [2]. The aim of the management in older children is to block hyperandrogenism and to prevent or manage the complications of the classic form and its treatment. The treatment may need to be completed by a gender-affirming surgery which constitutes a great challenge given the necessity of participation of a gynecologist and a pediatric surgeon in female patients [3]. Precocious puberty (PP) refers to the emergence of secondary sexual characteristics before the age of 8 in females and 9 in males; classified into central and peripheral PP depending on the presence or absence of premature activation of the hypothalamic-pituitary-gonadal axis, respectively [4].
We report in this article, a rare case of a 9-year-old male who has been diagnosed with 21-hydroxylase deficient CAH that presented with peripheral PP. This will certainly help to increase awareness among health care workers and highlight the importance of early diagnosis and treatment.
Case Report
Clinical Presentation
Our case is a 9-year-old boy who presented for the first time to our tertiary hospital at the age of 4. The Road to Health Booklet provided, showed that the patient was born via normal vaginal delivery at term and the perinatal history was uneventful. However, at the age of 4, he was found to be taller than most of his peers with a large penis and pubic hair. He has two siblings with normal health. There was no maternal history of hirsutism or genital abnormality and no family history of infertility or atypical genitalia or unexplained neonatal death. His neurodevelopment assessment was noted to have normal gross and fine motor skills. The speech and language capabilities were within normal range. Although he was assessed to have mild intellectual disability, he was able to attend a mainstream school. On clinical exam, his weight was 24.4 kg, height 122 cm, pulse rate 94 beats/minute, temperature 36.9 °C and blood pressure 106/49 mmHg. His body mass index plotted above the 97th percentile. On genital examination, Tanner stage IV male pattern pubic hair was noted, with a mature penis but appropriate prepubertal testes size and no gynecomastia.
Laboratory and Radiological Investigations
Laboratory investigations revealed serum electrolytes that were within reference intervals throughout. He had low follicle-stimulating hormone (FSH) and luteinizing hormone (LH) (<1.0 IU/L for both), high testosterone 12.5 nmol/L (0.1–0.9) and high 17-OH progesterone 87.0 nmol/L (0.5–2.2). Serum cortisol and plasma adrenocorticotropic hormone were not done.
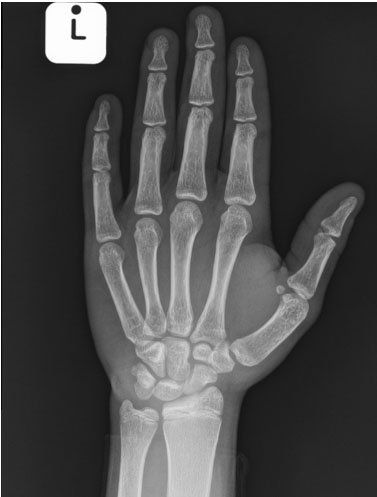
An X-ray of his left (non-dominant) hand revealed premature fusion of the epiphysis to metaphysis with closure of the physis in the distal phalanges, metacarpals, proximal and middle phalanges. The epiphysis of the distal radius and ulna were the same width as their corresponding metaphysis. The bone maturity of the patient was of late puberty stage in-keeping with advanced bone age of a 17-year-old boy (Figure 1).
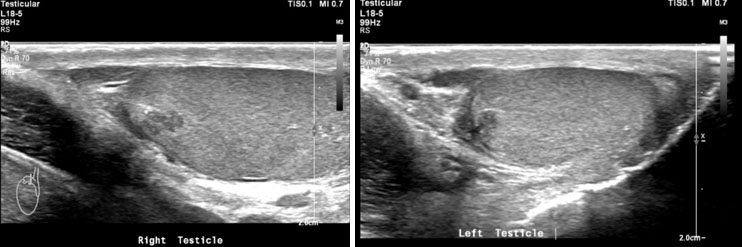
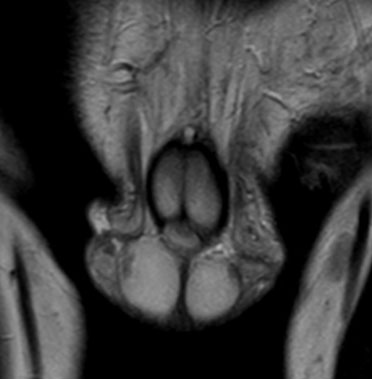
Adrenal rests were noted in both testes; the scrotal ultrasound and MRI showed bilateral eccentric lobulated lesions adjacent to the mediastinum in the testes. No calcification or vascularity was seen in the testicular lesions (Figure 2A, Figure 2B and Figure 3).
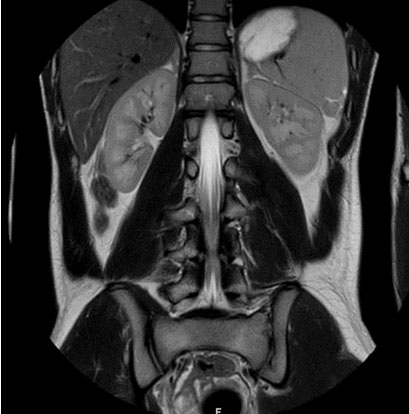
Computed tomography (CT) and magnetic resonance (MR) images of the adrenal glands demonstrated normal adrenal glands (Figure 4). There were no urinary tract malformations in imaging.
Confirmatory genetic testing was performed through the National Health Laboratory Service (NHLS), Inherited Metabolic Diseases Molecular Diagnostic Laboratory in Cape Town, South Africa. The utilization of multiplex ligation-dependent probe amplification (MLPA), followed by sequencing of the CYP21A2 gene revealed compound heterozygosity for c.293-13C>G splice site variant and c.955G>A p.(Gln319Ter).
Management and Progress
After counselling the patient and family, he was started on prednisone 5 mg daily and fludrocortisone 100 mcg po daily. The repeat investigations at three months on treatment showed a marked decrease in 17-OH progesterone to 37.3 nmol/L and testosterone to 9.2 nmol/L. The blood pressure, serum electrolytes and parameters of renal function were still maintained within reference ranges.
Discussion
We have reported here on a male child presenting as peripheral PP due to CAH. A 21-hydroxylase deficiency accounts for approximately 95% of CAH cases [5]. The classic CAH due to 21-hydroxylase deficiency, comprises of two forms, salt-wasting (SW-CAH) and simple virilizing (SV-CAH). The simple virilizing (SV) type is characterized by cortisol deficiency and androgen excess with sufficient aldosterone synthesis supposedly due to milder enzyme deficiency [6],[7]. Most classic CAH cases present with SW and only 25% of affected patients present with the SV type [8].
The hallmark of the SV-CAH is virilization in females and precocious puberty in males along with enhanced somatic growth; all due to androgen hypersecretion. The index patient presented with features of peripheral precocious puberty, secondary sexual characteristics, and advanced bone age with no clinical and biochemical features of SW. The revelation of high testosterone level with low FSH and LH associated with pre-pubertal testes size are indicative of peripheral PP in our case [9]. Congenital adrenal hyperplasia is associated with adrenal abnormalities, namely, adrenal hyperplasia, myelolipoma, benign adenoma, and rarely pheochromocytoma [10]. In patients with CAH, myelolipoma is common in those who were diagnosed late or poorly controlled. Although, no adrenal abnormalities were noted in our patient, follow-up imaging of patients with CAH is important especially in those who present with abdominal pain [11]. Male patients may have ectopic adrenal tissue which may be found anywhere along the gonadal descent pathway [10]. The reported prevalence of testicular adrenal rests in patients with CAH is 24% [12]. Classically, the ectopic adrenal tissue in males may be found in the testes. Likewise, testicular adrenal rests were noted in both testes of the index case.
In utero virilization seldom occurs in males, instead males with SV-CAH present with clinical features of androgen excess at 2–4 years, the age at which our patient presented for the first time at our institution. It is through newborn screening (NBS) that milder forms of CAH particularly in males can be detected early in the neonatal period [13]. However, the timing of sample collection is crucial because a relatively high proportion of cases with SV-CAH are reported to be missed if a single 17-OH progesterone measurement is done soon after birth [14],[15]. Few case reports of CAH in males have been published. Ruma Parvin et al. reported on a male neonate with SW-CAH that responded well to early treatment [2]. The mortality rate is reported to be higher in males as a result of males not being detected early in the neonatal period particularly in developing countries [6]. In addition to high mortality due to adrenal crises, the undiagnosed male infants endure the morbidity associated with early adrenarche and growth acceleration [16].
In a long term, retrospective South African study, the female participants (80.8%) were considerably higher than the males (19.2%). All the male participants were diagnosed with SW-CAH after presenting with dehydration and shock. As expected, the diagnoses of SV-CAH were established significantly later at a median age of 8.1 years whilst the SW-CAH was diagnosed at a median age of 2 months [7]. Two case reports have highlighted the diagnostic challenges of SV-CAH, demonstrated by late diagnoses due to initial 17-OH progesterone results that were within the reference ranges [8],[17] Joo, Yoo et al. 2024 reported that the diagnosis was made after 17-OH progesterone elevation post adrenocorticotrophic hormone (ACTH) stimulation test which is the gold standard for non-classic CAH [17]. Majumdar, Sharan et al. 2017 have published a case report of 6-year-old male with CAH presenting initially as peripheral PP which subsequently converted to central PP as a result of prolonged exposure to androgens [9]. The late diagnosis of CAH predisposes patients to uncontrolled adrenal androgen secretion which may complicate as central PP. Conversely, vigorous glucocorticoid treatment is capable of inducing a sudden contraction in androgen levels, thereby causing centrally mediated PP due to secretion of the pituitary gonadotropins particularly in late onset CAH [17].
Confirmatory genetic testing for CAH and other inherited metabolic diseases is seldom considered and done in South Africa even though testing is available through the NHLS. Identification of biallelic pathogenic variants in CYP21A2 gene confirms the diagnosis of CAH and permits for accurate genetic counselling and family studies in the form of carrier and prenatal testing. The pathogenic variants detected in our patient have been previously reported. The c.293-13C/A>G (In2G) variant is regarded as the most common splicing variant in CYP21A2 gene typically leading to SW-CAH in numerous ethnicities among different countries [18]. Regarding the c.955G>A p.(Gln319Ter) variant, the glutamine amino acid is being replaced by a premature stop codon leading to a truncated and non-functional enzyme. It has been reported that heterozygosity for p.(Gln319Ter) is associated with milder non-classical forms of CAH, which may explain the phenotype in our patient [19].
The unavailability of universal NBS in South Africa contributes to treatable conditions such as CAH being missed leading to potentially dire consequences to affected patients and their families. The mortality rate in SW-CAH without screening is in the range of 4–10% [7]. Meanwhile, patients with SV-CAH are also at risk of complicating with adrenal crisis as a result of cortisol deficiency. In the absence of NBS, higher standards of clinical awareness and practice will be imperative for early intervention to achieve desired clinical outcomes. The ongoing challenges of diagnosis and adequate management in under-resourced settings like ours negatively impact access to care and the reporting of CAH prevalence in South Africa. This is further aggravated by some cultural, religious and social issues that may arise from the clinical presentation of atypical genitalia, and the impact this may have on overall patient care and treatment adherence.
Conclusion
There are limited published data on CAH in male infants in South Africa. The current case study highlights the importance of a thorough history taking and clinical examination at birth, appropriate investigations including radiological imaging and early management and/or referral of any anomaly of sexual development. The utility of molecular testing in attaining definitive diagnoses should be encouraged in our setting in order to offer accurate genetic counselling to the families and assisting them with planning for future pregnancies.
REFERENCE
1.
El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet 2017;390(10108):2194–210. [CrossRef]
[Pubmed]

2.
Parvin R, Ghosh NK, Mahbuba S, Chudhury FJ, Ferdoucy SA. A male neonate with congenital adrenal hyperplasia: A case report. DS (Child) HJ 2020;36(1):75–7. [CrossRef]
3.
Alla A, Draoui N, Rami I, Rouf S, Saadi H, Kamaoui I, et al. A rare case report about a congenital adrenal hyperplasia by 21-hydroxylase lock in its pure virilizing form discovered in adolescence. Ann Med Surg (Lond) 2022;78:103673. [CrossRef]
[Pubmed]
4.
Berberoğlu M. Precocious puberty and normal variant puberty: Definition, etiology, diagnosis and current management. J Clin Res Pediatr Endocrinol 2009;1(4):164–74. [CrossRef]
[Pubmed]
5.
Dallos-Lara MF, Mendoza-Rojas VC. Pubertad precoz por hiperplasia adrenal congénita. Reporte de caso/Precocious puberty due to congenital adrenal hyperplasia. Case report. Rev Fac Med 2020;68(1).
6.
Grosse SD, Van Vliet G. How many deaths can be prevented by newborn screening for congenital adrenal hyperplasia? Horm Res 2007;67(6):284–91. [CrossRef]
[Pubmed]
7.
Ganie Y, Aldous C, Balakrishna Y, Wiersma R. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency in South Africa. S Afr Med J 2018;108(2):132–7. [CrossRef]
[Pubmed]
8.
Singh R, Agarwal M, Sinha S. Challenges in the diagnosis of simple-virilizing congenital adrenal hyperplasia: A case report. Cureus 2022;14(10):e29966. [CrossRef]
[Pubmed]
9.
Majumdar J, Sharan A, Mukhopadhyay S, Ghosh B, Sengupta S. Congenital adrenal hyperplasia—A rare cause of central precocious puberty—A case report. Journal of Clinical and Diagnostic Research 2017;11(11):SD01–3. [CrossRef]
10.
Murphy A, Kearns C, Sugi MD, Sweet DE. Congenital adrenal hyperplasia. Radiographics 2021;41(4):E105–6. [CrossRef]
[Pubmed]
11.
Nermoen I, Falhammar H. Prevalence and characteristics of adrenal tumors and myelolipomas in congenital adrenal hyperplasia: A systematic review and meta-analysis. Endocr Pract 2020;26(11):1351–65. [CrossRef]
[Pubmed]
12.
Kok HK, Sherlock M, Healy NA, Doody O, Govender P, Torreggiani WC. Imaging features of poorly controlled congenital adrenal hyperplasia in adults. Br J Radiol 2015;88(1053):20150352. [CrossRef]
[Pubmed]
13.
Thil’en A, Nordenström A, Hagenfeldt L, von Döbeln U, Guthenberg C, Larsson A. Benefits of neonatal screening for congenital adrenal hyperplasia (21-hydroxylase deficiency) in Sweden. Pediatrics 1998;101(4):E11. [CrossRef]
[Pubmed]
14.
Votava F, Török D, Kovács J, Möslinger D, Baumgartner-Parzer SM, Sólyom J, et al. Estimation of the false-negative rate in newborn screening for congenital adrenal hyperplasia. Eur J Endocrinol 2005;152(6):869–74. [CrossRef]
[Pubmed]
15.
Varness TS, Allen DB, Hoffman GL. Newborn screening for congenital adrenal hyperplasia has reduced sensitivity in girls. J Pediatr 2005;147(4):493–8. [CrossRef]
[Pubmed]
16.
Perry R, Kecha O, Paquette J, Huot C, Van Vliet G, Deal C. Primary adrenal insufficiency in children: Twenty years experience at the Sainte-Justine Hospital, Montreal. J Clin Endocrinol Metab 2005;90(6):3243–50. [CrossRef]
[Pubmed]
17.
Joo EY, Yoo MJ, Kim SJ, Jang W, Lee JE. Case report: Development of central precocious puberty in a girl with late-diagnosed simple virilizing congenital adrenal hyperplasia complicated with Williams syndrome. Front Endocrinol (Lausanne) 2024;15:1352552. [CrossRef]
[Pubmed]
18.
Kocova M, Concolino P, Falhammar H. Characteristics of In2G variant in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Front Endocrinol (Lausanne) 2022;12:788812. [CrossRef]
[Pubmed]
19.
Fanis P, Skordis N, Toumba M, Picolos M, Tanteles GA, Neocleous V, et al. The pathogenic p.Gln319Ter variant is not causing congenital adrenal hyperplasia when inherited in one of the duplicated CYP21A2 genes. Front Endocrinol (Lausanne) 2023;14:1156616. [CrossRef]
[Pubmed]
SUPPORTING INFORMATION
Author Contributions
Tumelo M Satekge - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Jean Paul Mulang Tshipeng - Conception of the work, Design of the work, Acquisition of data, Drafting the work, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Refiloe Johana Khosa - Analysis of data, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Chris Sutton - Conception of the work, Design of the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guaranter of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthors declare no conflict of interest.
Copyright© 2025 Tumelo M Satekge et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.



